
 Login
Login CN
CN

 2025.12.04.
2025.12.04.
Huntington's disease is caused by a dominant mutation in the first exon of the Huntingtin (HTT) gene on chromosome 4. Specifically, it involves the expansion of the CAG trinucleotide repeat sequence beyond 35 repetitions. This leads to an abnormal elongation of the encoded polyglutamine (polyQ) chain, forming a mutant Huntingtin protein (mHTT). mHTT accumulates abnormally within neurons, forming inclusions that disrupt cellular functions and ultimately lead to neuronal death. Research has shown that a higher number of CAG repeats is associated with an earlier age of onset and more severe disease progression.
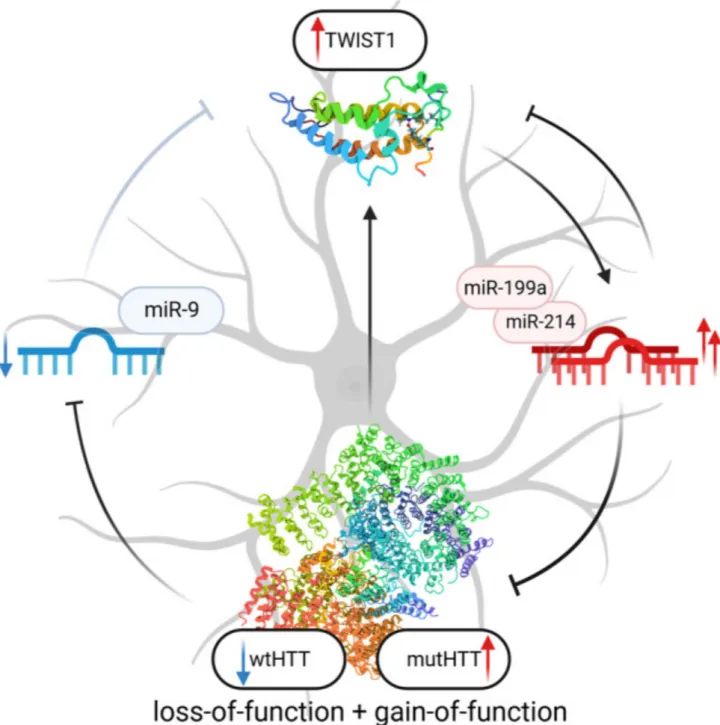
Image source: pubmed
Gene Therapy
Antisense Oligonucleotide (ASO) Therapy: This approach involves using ASOs to specifically bind to mRNA, preventing the synthesis of mHTT. Recent studies have found that the DNA mismatch repair protein MSH3 drives the somatic expansion of CAG repeats. ASOs targeting MSH3 effectively reduced CAG repeat expansion in striatal neurons derived from induced pluripotent stem cells (iPSCs), demonstrating a good safety profile.
Gene Editing Technology: This technology can directly edit the HTT gene to correct the mutation. A team from the Guangdong-Hong Kong-Macau Institute of CNS Regeneration at Jinan University pioneered the application of a gene editing system in a large animal model of Huntington's disease (a pig model), successfully and efficiently knocking down HTT mRNA. In a Huntington's disease 140Q-KI mouse model, this technique attenuated neuroinflammation and improved motor dysfunction.
Furthermore, a team led by Cai Yujia at Shanghai Jiao Tong University, in collaboration with Fudan University and AstraZeneca, developed a novel gene editing delivery tool named RIDE (Ribonucleoprotein delivery). This technology utilizes virus-like particles (VLPs) to deliver ribonucleoprotein (RNP), enabling cell-specific gene editing in neuronal cells. In a mouse model of Huntington's disease, this approach significantly reduced mHTT expression and ameliorated disease symptoms by knocking out the CAG repeat sequence.
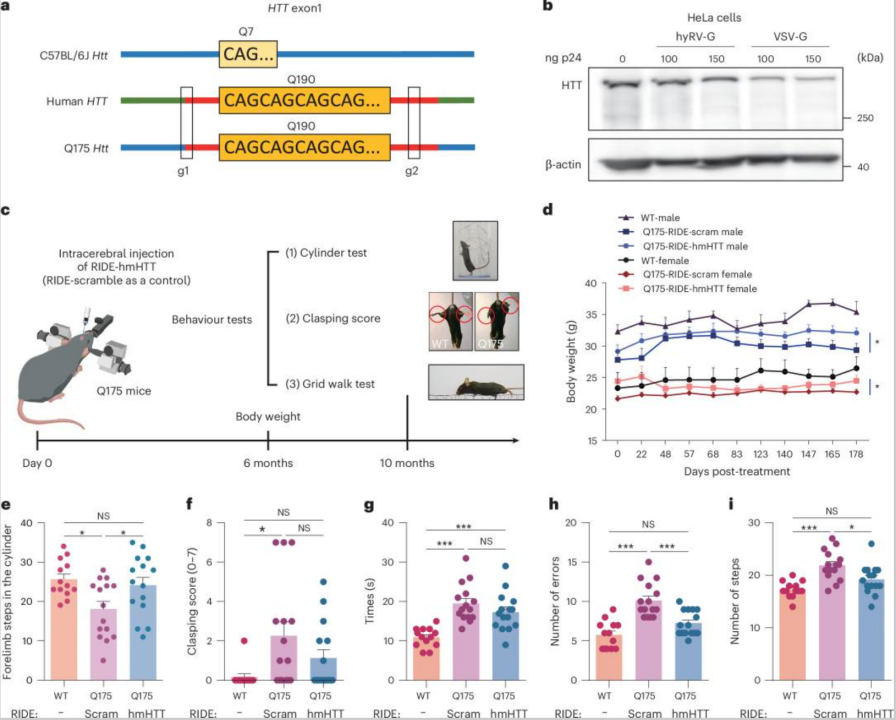
RNA Interference (RNAi) Technology: RNA interference utilizes small interfering RNA (siRNA) or short hairpin RNA (shRNA) to specifically degrade target mRNA. Artificially designed miRNAs can be engineered to specifically target HTT mRNA. In a transgenic sheep model, an artificial miRNA significantly reduced the levels of human mHTT throughout the striatum.
Mice model
HTT N-terminal gene editing model: Carries a small portion of the 5' end of the human HTT gene, including exon 1 containing the CAG repeat region.
R6/1 mouse: Contains 115 CAG repeats, with a slow onset of symptoms. Neurological symptoms appear at 22–26 weeks, and the terminal stage is reached at 38–40 weeks. Main manifestations include motor deficits, weight loss, and striatal degeneration.
R6/2 mouse: Contains 145 CAG repeats, with an early onset and rapid progression. Motor dysfunction, weight loss, and striatal volume reduction appear at 5–6 weeks, and the terminal stage is reached at 12–14 weeks. It is one of the most widely used models.
Full-length HTT gene editing model: Expresses full-length mutant human HTT protein via yeast or bacterial artificial chromosomes (YAC or BAC).
YAC128 mouse: Contains 128 CAG repeats. Hyperactivity is observed at 3 months, followed by motor deficits, and hypokinesia at 12 months. It can mimic slowly progressing HD.
BACHD mouse: Contains 97 mixed CAA-CAG repeats. It exhibits progressive motor deficits, neuronal synaptic dysfunction, and striatal atrophy, making it suitable for preclinical studies.
BAC226Q mouse: Carries 226 mixed CAG-CAA repeats and includes endogenous human HTT promoters and regulatory elements. It shows motor deficits, striatal atrophy, neuronal death, and shortened lifespan. Compared to BACHD, its symptoms are more accurate and onset is earlier. Motor deficits manifest as sudden twitching and twisting of the head and body, similar to the choreiform movements of HD patients.
Knock-in model: Replaces exon 1 of the mouse Htt gene with the human HTT CAG repeat sequence.
zQ175 mouse: Derived from spontaneous expansion of CAG repeats in the CAG140 mouse model. Motor deficits and weight loss appear at 2–4 months, and striatum-dependent cognitive deficits appear at 10–12 months.
HdhQ150 mouse: Significant motor deficits and weight loss begin at 70 weeks, and a reduction in striatal neuron count is observed at 100 weeks. Due to its prolonged neuronal functional decline, it is useful for studying the mechanisms of early neuronal degeneration in HD.
CAG140 mouse: Contains 140 CAG repeats. These mice exhibit increased activity at 4–5 weeks, followed by reduced activity at 18–20 weeks, gait abnormalities at 1 year, and impaired long-term recognition memory. The olfactory system shows significant abnormalities in the early stages of HD.
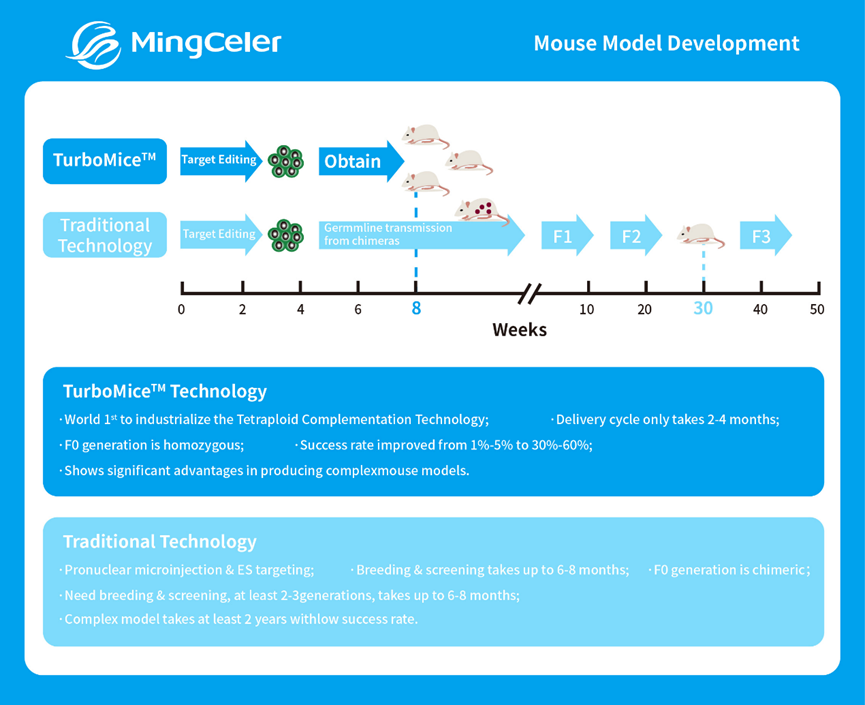
At Mingceler, we specialize in accelerating genetic research through our TurboMice™ technology, which harnesses optimized tetraploid complementation to deliver fully homozygous mouse models in just 2–4 months—60% faster than traditional breeding methods. Unlike conventional approaches that require 6–12 months of tedious breeding cycles, our process starts with precision editing of embryonic stem cells (ESCs) for targeted gene knockouts, floxing, or multi-locus modifications. We then use tetraploid complementation to develop these ESCs directly into experiment-ready mice, skipping the need for chimeric F1 and F2 generations. This method ensures 100% genotype certainty from the F0 generation, eliminating the guesswork of heterozygous screening and saving researchers thousands of hours spent on colony management.

